Introduction
Bulk RNA sequencing (RNAseq) experiments result in raw sequencing reads, typically in FASTQ format. In order to use this data to gain insights into gene expression patterns, identify novel transcripts, etc., it must undergo a series of processing steps – including quality control, read alignment, quantification of gene expression – and then further analysis depending on the aims of the experiment. Mantle can be used for data management, preprocessing, and downstream analysis of bulk RNAseq data. All data, pipelines, and analysis environments in this tutorial are available in your Mantle account.Raw data management
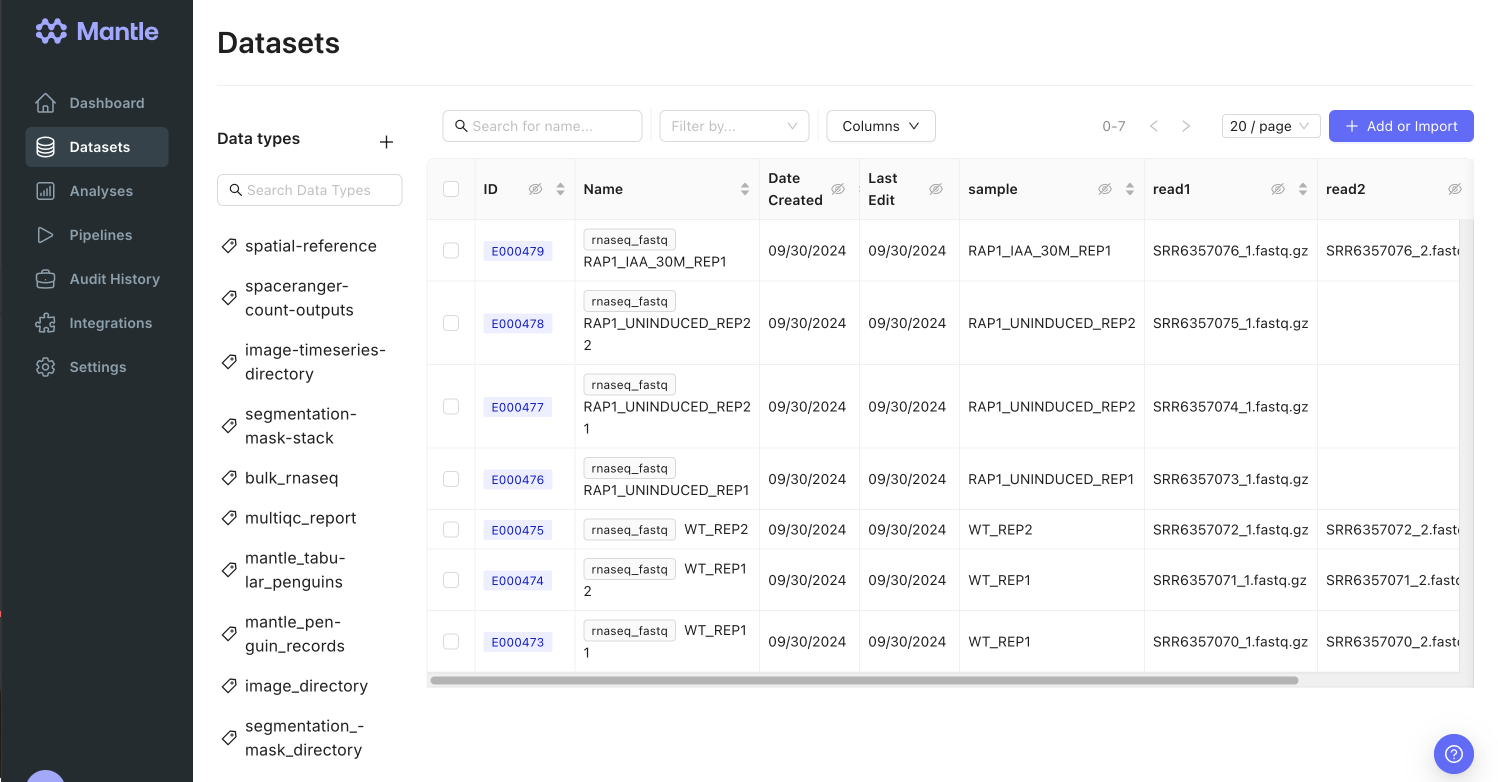
FASTQ data cannot be easily stored in generic databases. An individual FASTQ file is semi-structured, and storing FASTQ files within a database as binary large objects (BLOBs) can be challenging due to their large size (typically tens of gigabytes per FASTQ file). In Mantle, you can store data files and associated metadata together as datasets. Here, FASTQ files are stored as datasets of thernaseq_fastq data type. Expand to view the properties of rnaseq_fastq datasets:
Properties
Properties
Wu, A. C. K., Patel, H., Chia, M., Moretto, F., Frith, D., Snijders, A. P., & van Werven, F. J. (2018). Repression of divergent noncoding transcription by a sequence-specific transcription factor. Molecular Cell, 72(6), 942-954.e7. https://doi.org/10.1016/j.molcel.2018.10.018Additionally, in order to perform alignment, a reference genome FASTA file and gene annotation GTF or GFF file must be provided. Additional reference data, such as a reference transcriptome or additional FASTA could be required depending on your analysis. We’ve used the
rnaseq_reference data type to allow you to store all reference data together. Expand to view the properties of rnaseq_reference datasets:
Properties
Properties
Where did the reference data come from — e.g. ENSEMBL
The name of the genome and/or other identifying information for the reference dataset
Species of the genome (optional)
Genome FASTA file
GTF annotation file
GFF annotation file (optional — specify if you don’t have a GTF file)
Transcriptome FASTA file (optional)
Specify if your GTF annotation is in GENCODE format (optional — if your GTF file is in GENCODE format and you would like to run Salmon i.e. —pseudo_aligner salmon, you will need to provide this parameter in order to build the Salmon index appropriately)
The attribute type used to group features in the GTF file when running Salmon (optional)
The attribute type used to group feature types in the GTF file when generating the biotype plot with featureCounts (optional)
By default, the pipeline assigns reads based on the ‘exon’ attribute within the GTF file (optional)
FASTA file to concatenate to genome FASTA file e.g. containing spike-in sequences (optional)
Data processing
Bulk RNAseq FASTQs need to be preprocessed to obtain count matrix or other forms of data that can be analyzed to draw insights. This can be accomplished using the nf-corernaseq pipeline, which is included in your Mantle account.
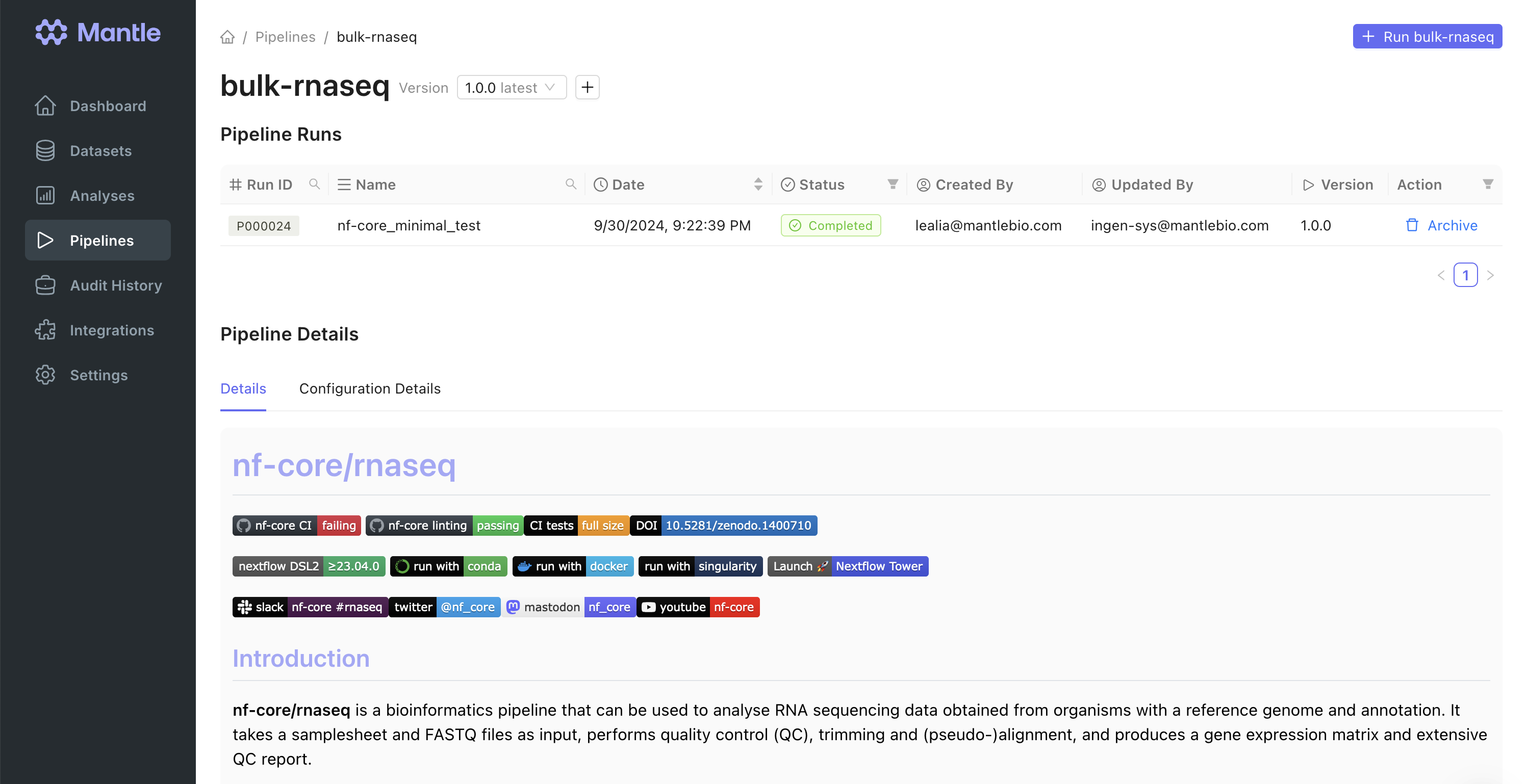
rnaseq_fastq datasets into an input samplesheet. Additionally, we added a process at the end of the workflow to collect some of the files that the pipeline creates into Mantle datasets, and to register the MultiQC report HTML file as an output of the pipeline on Mantle, which allows it to be displayed on the pipeline run page.
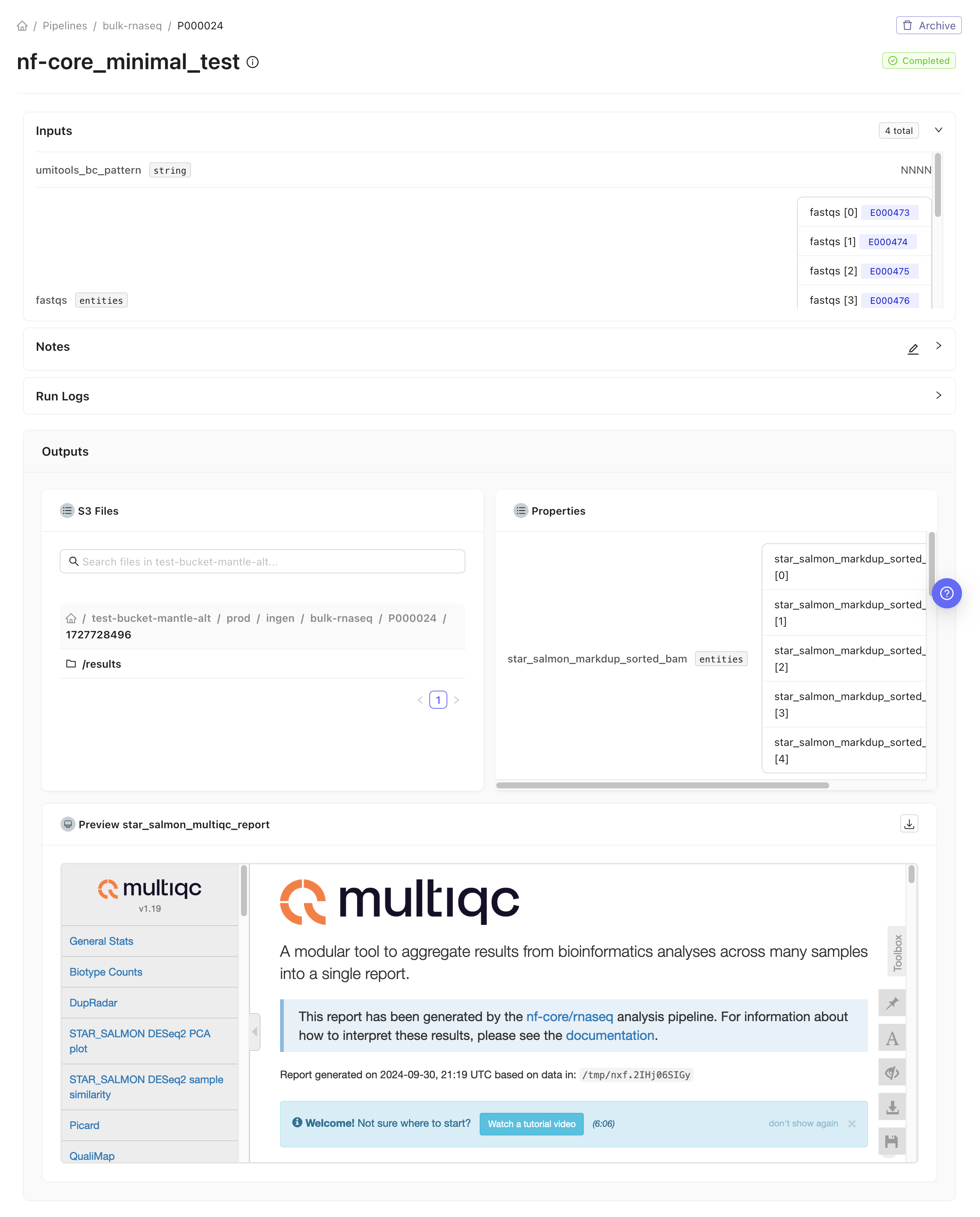
Analyzing processed data
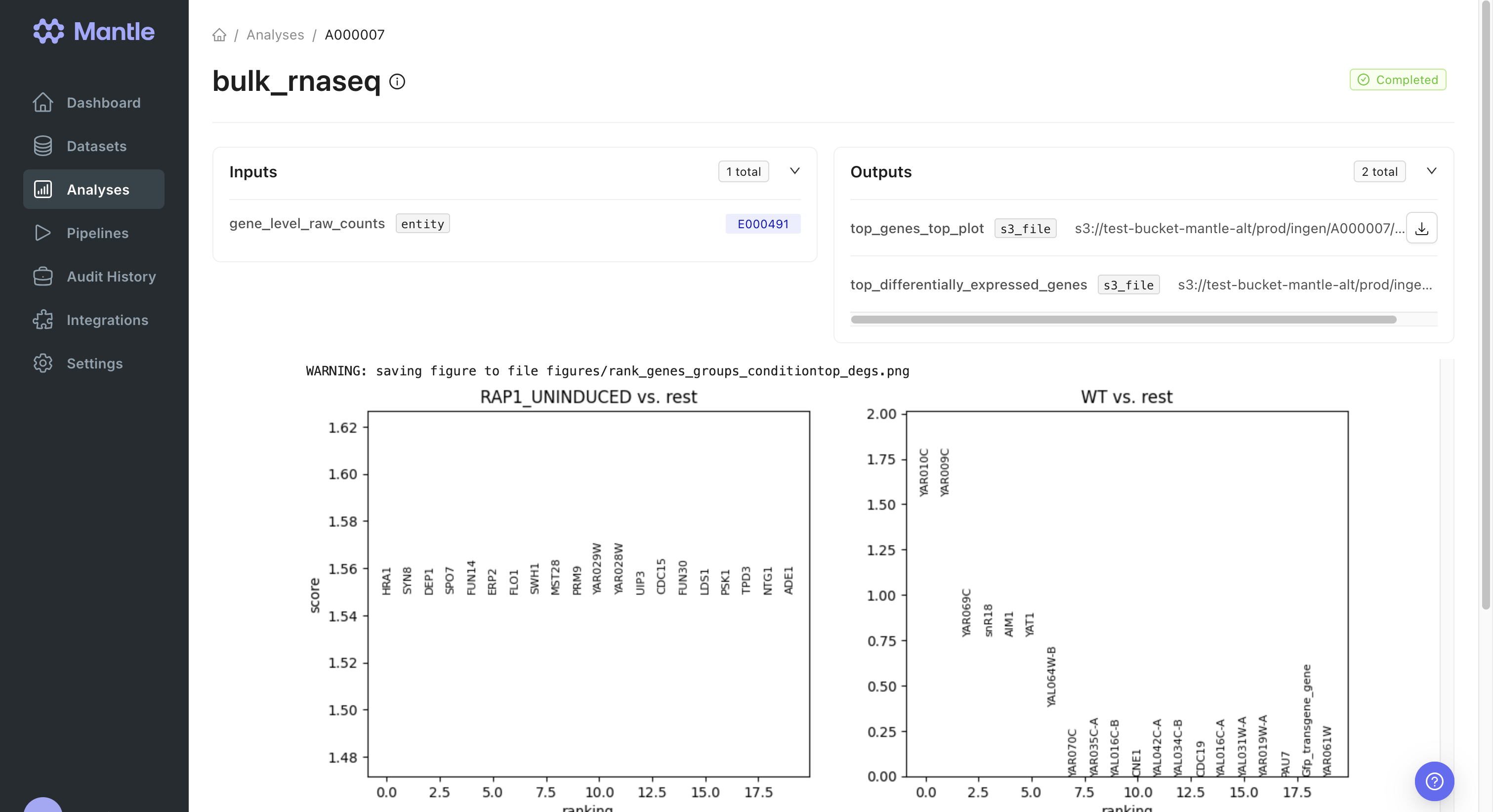
We analyzed data contained in the gene-level raw counts matrix produced by the pipeline (stored as acount_matrix dataset) in the bulk_rnaseq notebook, which ran in the spatial-transcriptomics-analysis environment.
matrix property on the dataset and mark the dataset as an input to the analysis notebook. Then, we used the Scanpy package to perform differential expression analysis.