Introduction
Spatial transcriptomics is the visualization and quantification of gene expression in tissue sections, maintaining the spatial context of the tissue architecture. One technology for spatial transcriptomics is 10x Genomics Visium. It uses a slide with an array of spots, each containing barcoded oligonucleotides, to capture mRNA from tissue sections placed on the slide. The mRNA then undergoes high-throughput RNA sequencing. The raw results from Visium experiments include a microscopy image of the tissue slice on the Visium slide and sequencing reads in FASTQ format. In order to use this data to reveal insights into cellular microenvironments, cellular heterogeneity, and spatial relationships within tissue, the data require processing, including the typical steps taken for processing RNA sequencing reads, as well as tissue detection and determination of which barcode spots overlap with the tissue. Mantle can be used for data management, preprocessing, and downstream analysis of spatial transcriptomics data. All data, pipelines, and analysis environments in this tutorial are available in your Mantle account.Raw data
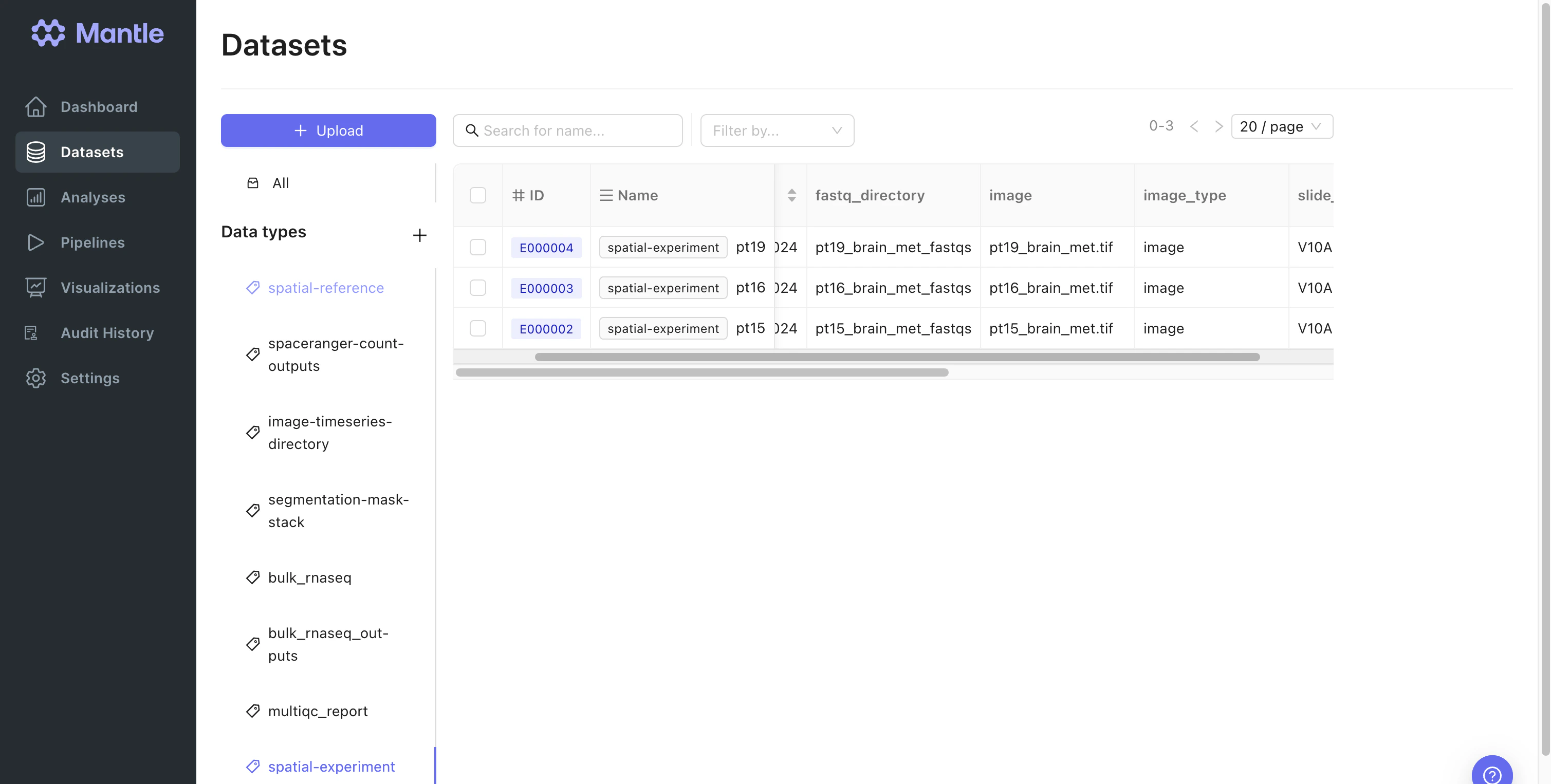
Both images and FASTQ data cannot be easily stored in typical databases. An individual FASTQ file is semi-structured, and images are unstructured. Storing these files within a database as binary large objects (BLOBs) can be challenging due to their large size (typically tens of gigabytes per FASTQ directory). In Mantle, you can store data files and associated metadata together as datasets. Here, experimental data files are stored as datasets of thespatial-experiment data type. Expand to view the properties of spatial-experiment datasets:
Properties
Properties
Directory containing RNA sequencing FASTQ files
Microscopy image file
image, darkimage, colorizedimage, or cytaimage.Used as an input to Spaceranger Count as --<image_type> <image>.See the Spaceranger Count documentation for more information.The Visium slide serial number. Corresponds to the
slide input to Spaceranger Count. See the Spaceranger Count documentation for more information.Optional. The slide design file for your slide. See the Spaceranger Count documentation for more information.
Optional. The slide design file for your slide. Corresponds to the
loupe-alignment input to Spaceranger Count. See the Spaceranger Count documentation for more information.Visium capture area identifier. Corresponds to the
area input to Spaceranger Count. See the Spaceranger Count documentation for more information.Optional. The publication that the data originated from.
Optional. The SRA Accession Number for the FASTQ files.
Sudmeier, et al. (2022) “Distinct phenotypic states and spatial distribution of CD8+ T cell clonotypes in human brain metastases.” Cell Reports Medicine, 3(5), 100620
GRCH38-2020-A) and mouse (mm10-2020-A) genomes. We packaged them into the spatial-reference datasets.
Depending on your experimental workflow, you may also need a spatial probe file for data processing. 10x also has probesets available for human and mouse, which we have packaged into the spatial-probes datasets.
Data processing
In our demonstration workflow, spatial transcriptomics experiment data is processed using a Nextflow pipeline that runs 10x Spaceranger Count. Spaceranger Count is a command line tool. Its inputs include FASTQ files, a microscope image, slide ID and capture area, and reference transcriptome. It is computationally intensive to run, requiring at least an 8-core processor (32 cores recommended), 64 GB RAM (128 GB recommended), and 1 TB free disk space. Ourspaceranger-count pipeline takes as input a dataset of the spatial-experiment type and a dataset of the spatial-reference type. The spatial-experiment dataset contains most of the Spaceranger Count required inputs as properties. The reference transcriptome is stored in the spatial-reference dataset. When you run this pipeline on Mantle, all the hardware provisioning is taken care of for you.
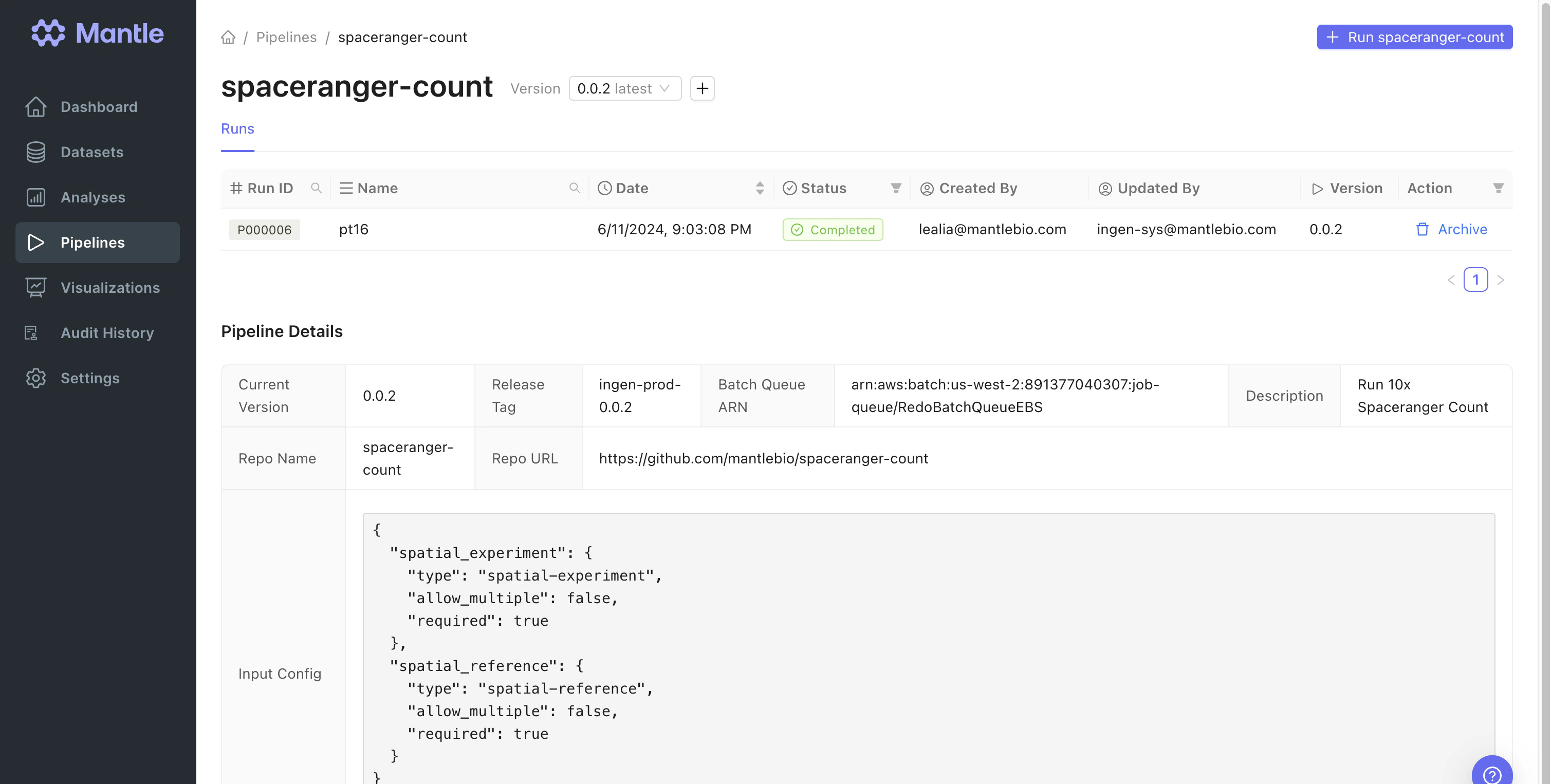
spaceranger-count pipeline outputs all the files generated by Spaceranger Count, and a spaceranger-outputs dataset. The spaceranger-outputs data type has
path_to_output_dir as a property, which contains the path to the directory containing the Spaceranger Count outputs.
The output dataset is used as an input to a downstream analysis notebook, and serves to link the Spaceranger Count outputs to the input datasets.
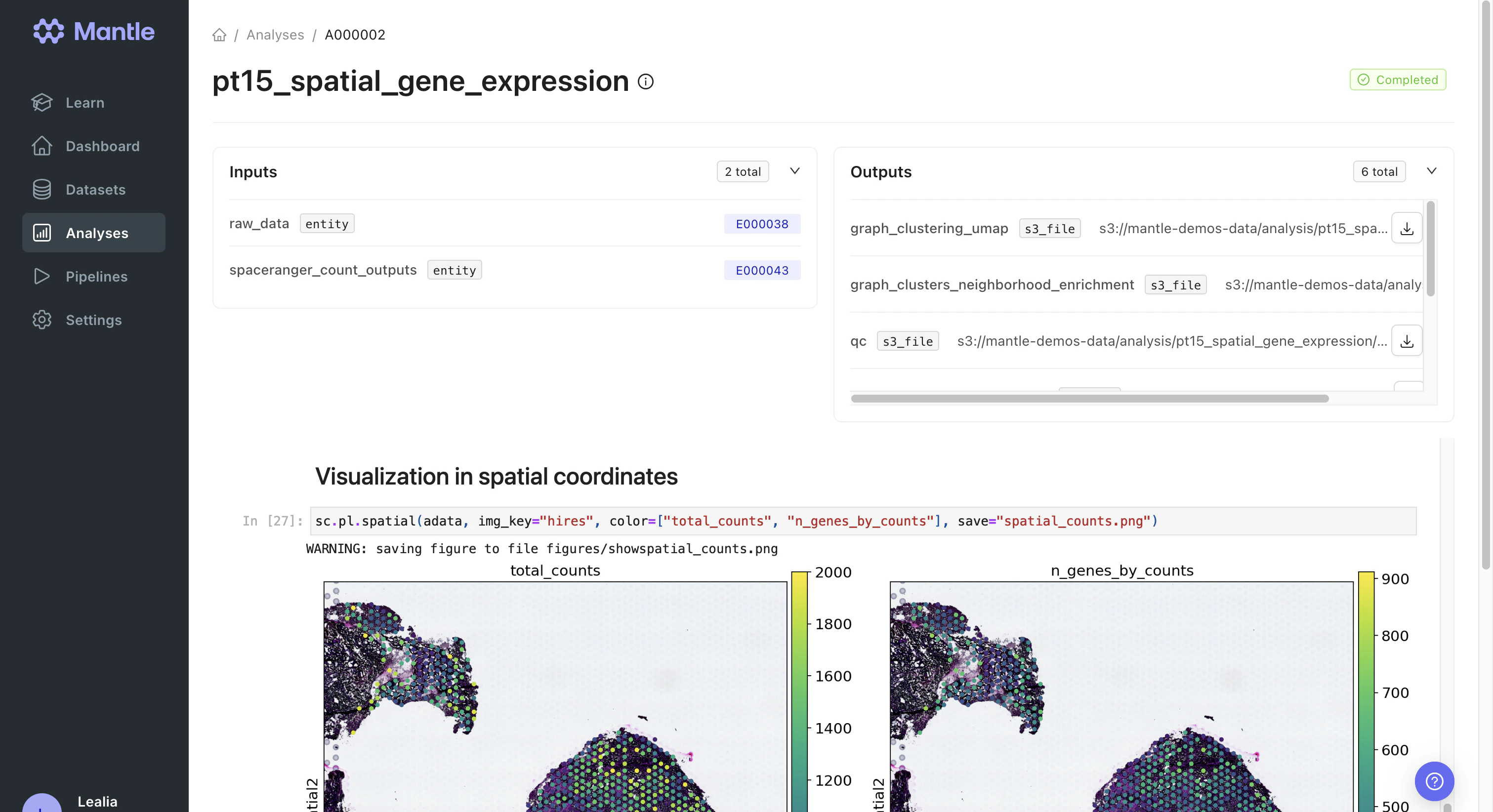
Analyzing processed data
We analyzed the Spaceranger Count outputs in thept15_spatial_gene_expression notebook, which ran in the spatial-transcriptomics-analysis analysis environment.