Introduction
A bioinformatics pipeline is a program used to process large biological data sets, such as sequencing data. It automates data workflows, enabling reproducibility and scalability in analyses. You can run pipelines on Mantle using a user-friendly graphical interface. The pipeline runs on powerful hardware in the cloud, so you don’t have to worry about downloading anything to your computer or running out of resources on your computer. Mantle pipelines are written in Nextflow , a workflow definition system with a large open-source bioinformatics pipeline development community, nf-core.Running a pipeline
1
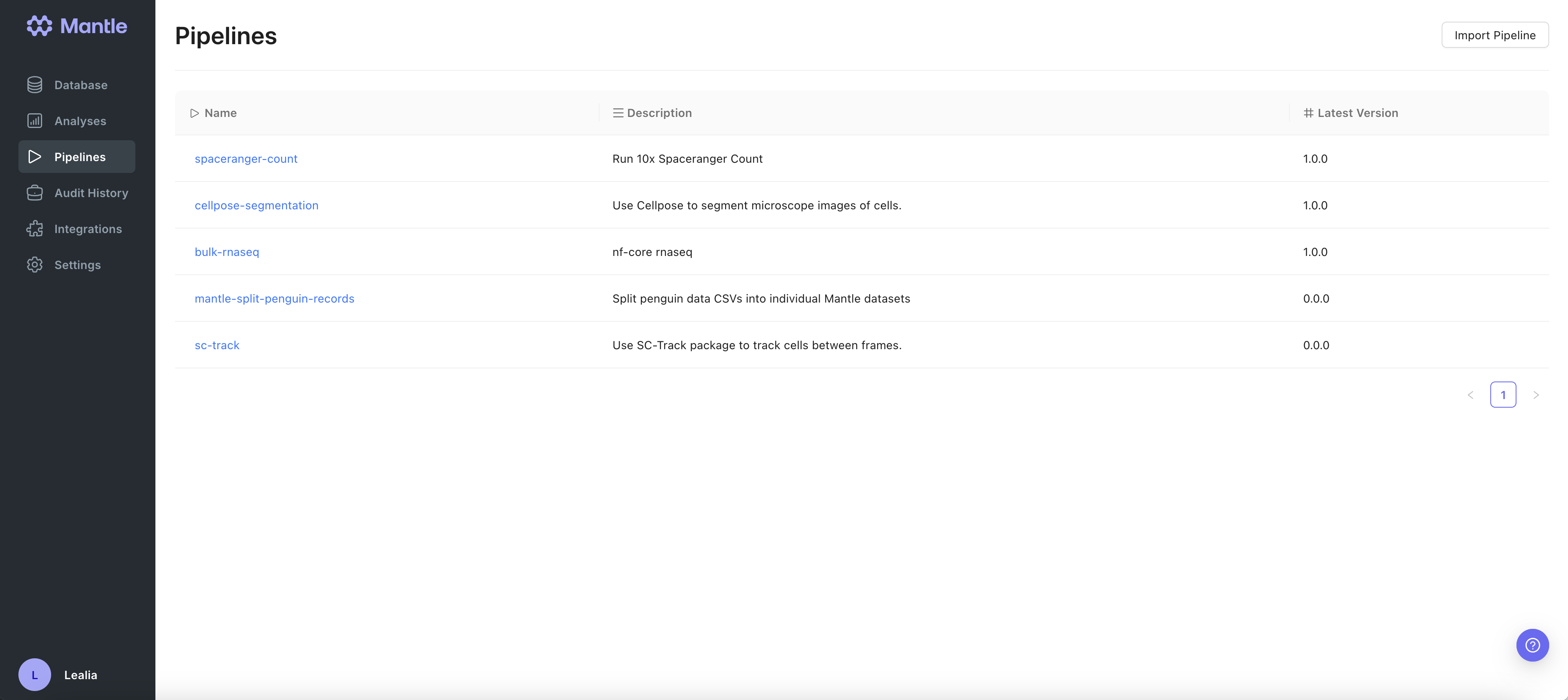
Navigate to the Pipelines tab within Mantle
2
Select the pipeline you want to run
3
Any previous runs are displayed on in the table on the pipeline’s page.
In addition, a README file is displayed at the bottom of the pipeline’s page.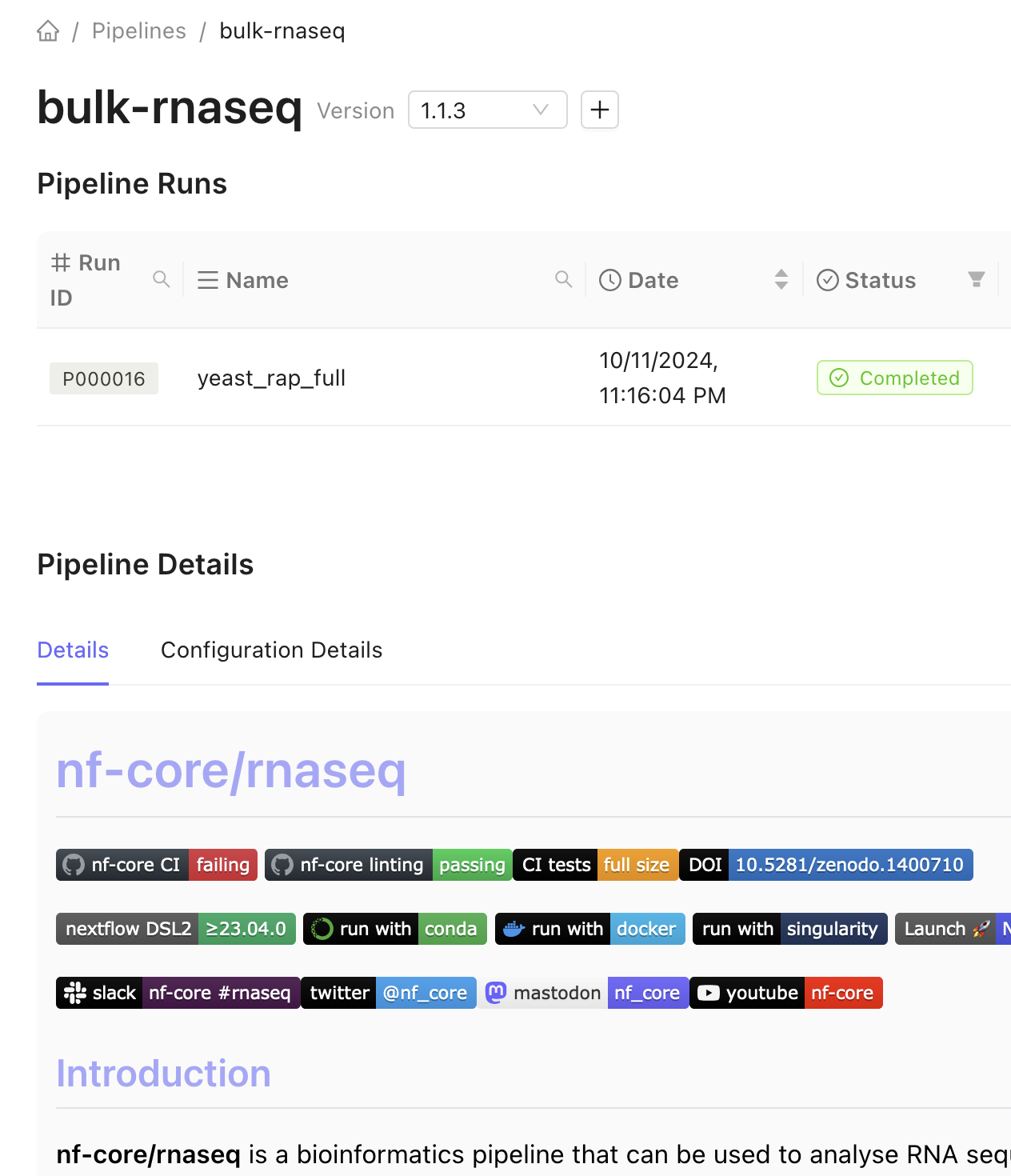
4
In the top right corner, click the + Run button.
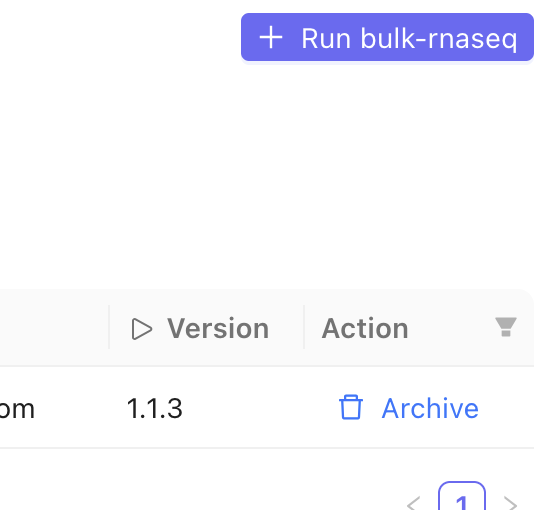
5
Fill out the form that appears and click the Run button
Mantle Pipelines are designed to take entities from your Mantle Database as inputs. For example, the bulk_rnaseq pipeline takes 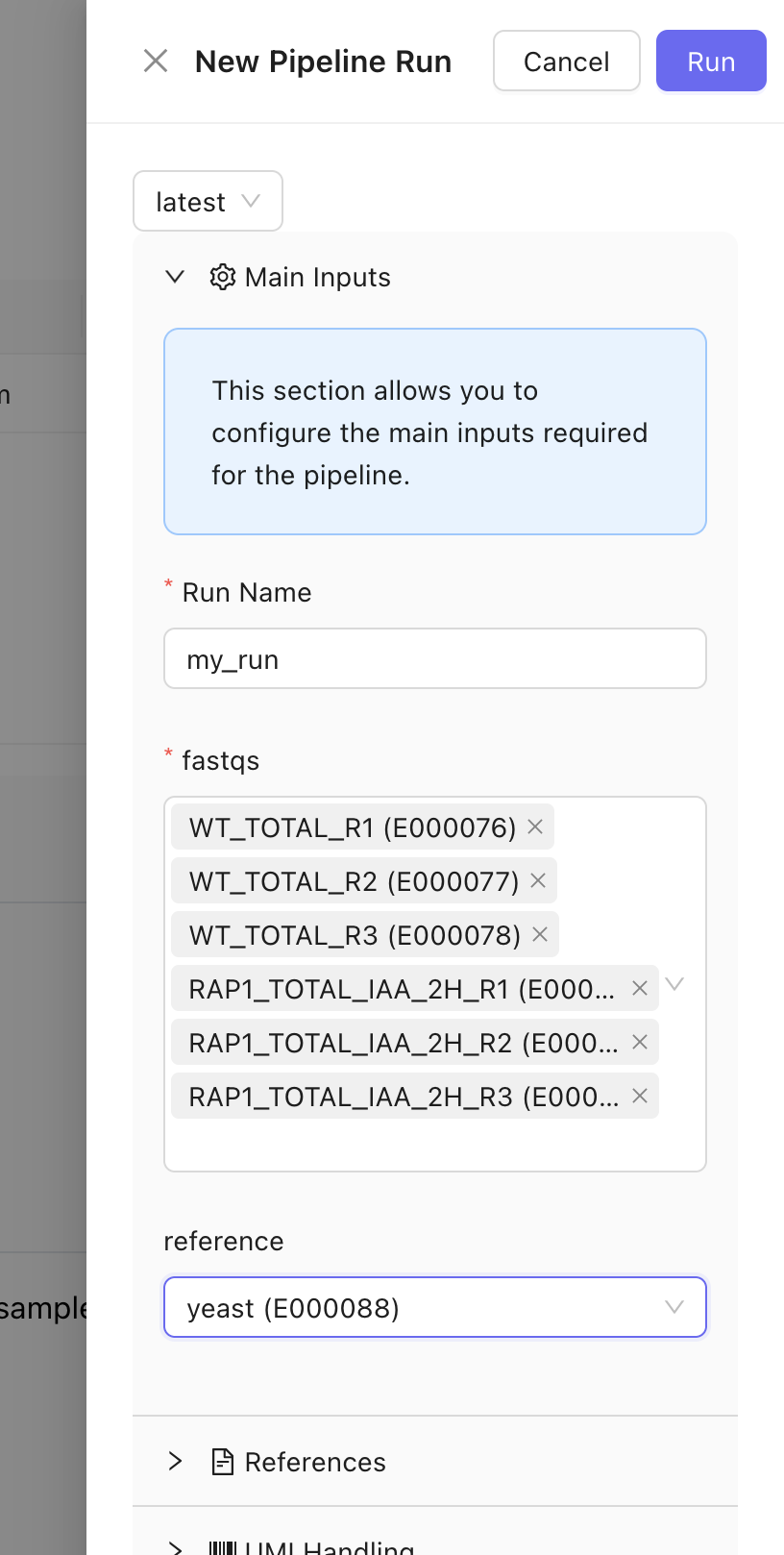
rnaseq_fastq entities and a rnaseq_reference entity in addition to string and number parameters.Accessing pipeline run outputs
On the Pipelines page
1
Click on the run you are interested in seeing the outputs of
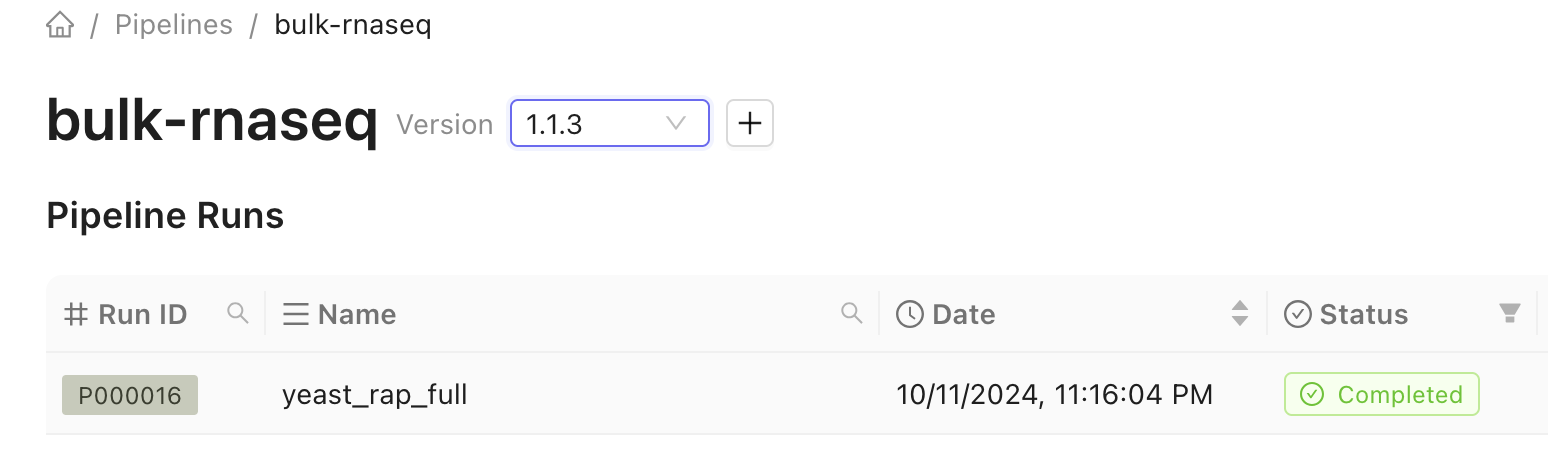
2
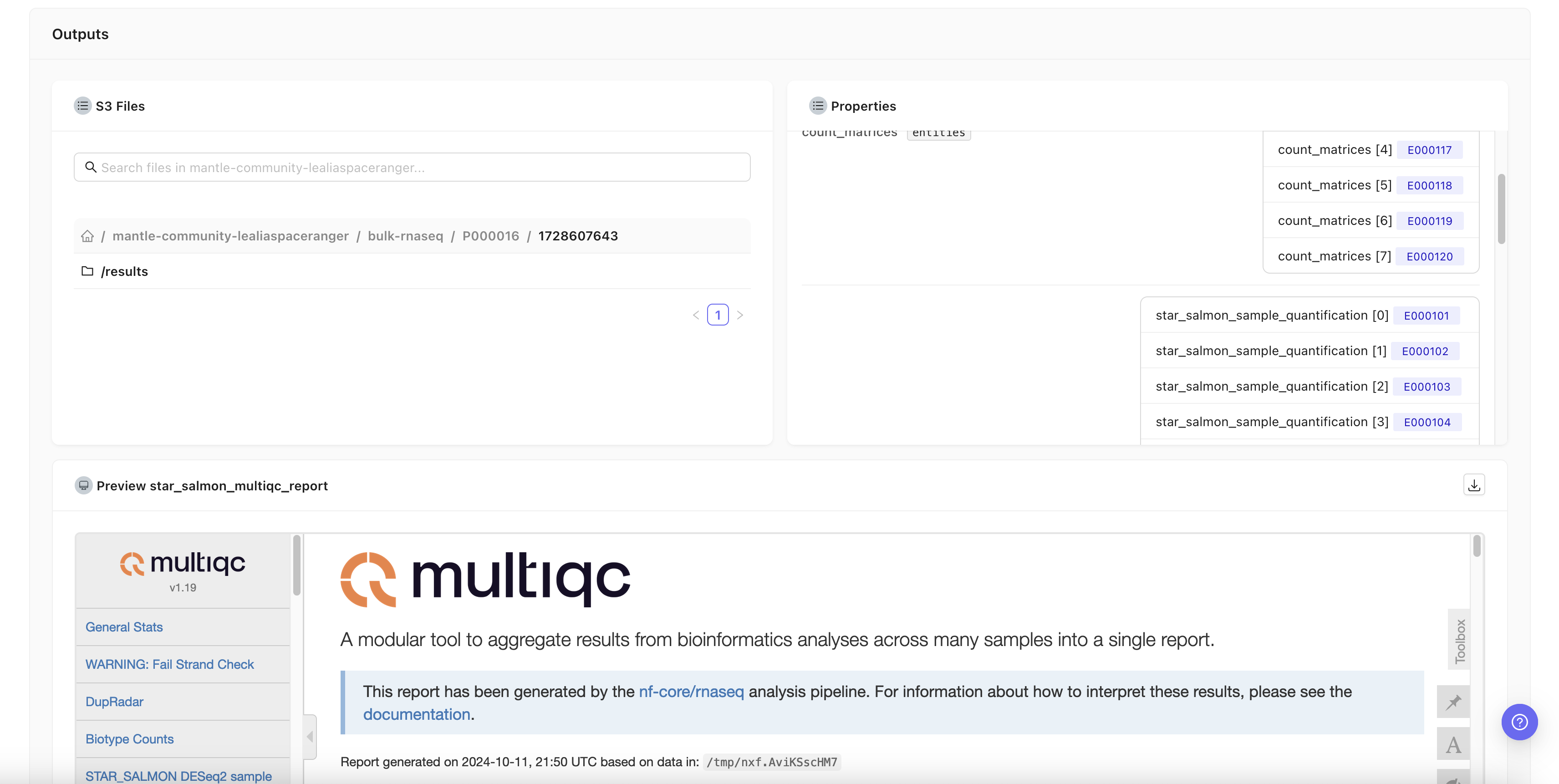
View results
All files that were output by the pipeline run can be browsed in the S3 Files section. Structured outputs are available in the Properties section. Some pipelines may also display visual outputs, such as HTML reports or images.