Introduction
Bulk RNA sequencing (RNAseq) experiments result in reads, typically in FASTQ format. In order to use this data to gain insights into gene expression patterns, identify novel transcripts, etc., it must undergo a series of processing steps – including quality control, read alignment, quantification of gene expression – and then further analysis depending on the aims of the experiment. The open-sourcernaseq pipeline from nf-core enables the transformation of FASTQ files to gene and isoform count matrices using STAR, RSEM, HISAT2, or Salmon.
We have adapted the nf-core rnaseq pipeline to read and write data to your Mantle Database.
Managing bulk RNAseq reads and references
In your Mantle Database, data is stored as entities, which have properties that can be files or metadata. Bulk RNAseq data can be stored in Mantle as entities of thernaseq_fastq data type. Expand to view the properties of rnaseq_fastq entities:
Properties
Properties
Wu, A. C. K., Patel, H., Chia, M., Moretto, F., Frith, D., Snijders, A. P., & van Werven, F. J. (2018). Repression of divergent noncoding transcription by a sequence-specific transcription factor. Molecular Cell, 72(6), 942-954.e7. https://doi.org/10.1016/j.molcel.2018.10.018Additionally, in order to perform alignment, a reference genome FASTA file and gene annotation GTF or GFF file must be provided. We’ve provided the
rnaseq_reference data type to allow you to store all reference data together. Expand to view the properties of rnaseq_reference entities:
Properties
Properties
string
required
Where did the reference data come from — e.g. ENSEMBL
string
required
The name of the genome and/or other identifying information for the reference dataset
string
Species of the genome (optional)
file
required
Genome FASTA file
file
GTF annotation file
file
GFF annotation file (optional — specify if you don’t have a GTF file)
file
Transcriptome FASTA file (optional)
bool
Specify if your GTF annotation is in GENCODE format (optional — if your GTF file is in GENCODE format and you would like to run Salmon i.e. —pseudo_aligner salmon, you will need to provide this parameter in order to build the Salmon index appropriately)
string
The attribute type used to group features in the GTF file when running Salmon (optional)
string
The attribute type used to group feature types in the GTF file when generating the biotype plot with featureCounts (optional)
string
By default, the pipeline assigns reads based on the ‘exon’ attribute within the GTF file (optional)
file
FASTA file to concatenate to genome FASTA file e.g. containing spike-in sequences (optional)
Processing bulk RNAseq reads to produce gene expression matrices
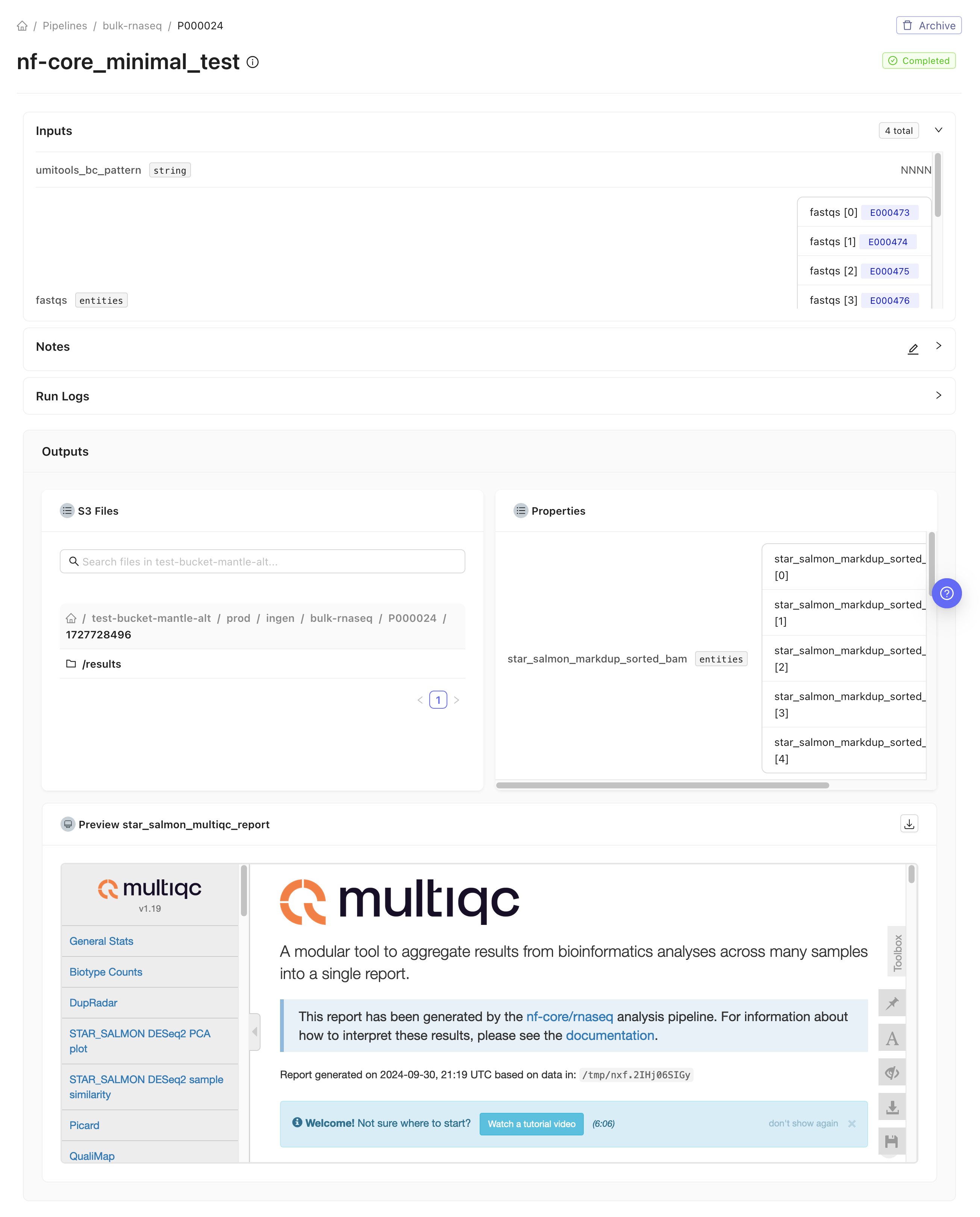
bulk-rnaseq pipeline adapts the nf-core rnaseq pipeline to allow it to take rnaseq_fastq entities as inputs (which it converts into the input samplesheet expected by the nf-core framework) and to collect some of the files that the pipeline creates into Mantle datasets. Additionally, when you run the Mantle bulk-rnaseq pipeline, the MultiQC report is displayed on the pipeline run page.
Some of the output entities include various gene expression matrices, such as raw gene counts matrix, and BAM files. The exact outputs depend on which alignment and quantification methods are specified to be used in the pipeline run.
All pipeline results files, including those not output to your Mantle Database, can be browsed on the pipeline run page. Learn more about the pipeline results files in the nf-core rnaseq documentation.